 Lecture
Lecture

8 y/o with three weeks of progressive edema of face, legs, and abdomen
Patient will present as → a 6-year-old boy who is brought to the emergency department by his mother due to swelling around his eyes and legs. The mother reports that the patient recently recovered from an upper respiratory tract infection. Physical exam is significant for periorbital and lower extremity edema. Laboratory testing is significant for hypoalbuminemia and normal complement levels. Urinalysis demonstrates 4+ protein and fatty casts with a "Maltese cross" sign.

To watch this and all of Joe Gilboy PA-C's video lessons you must be a member. Members can log in here or join now.
Nephrotic syndrome ⇒ patient has peripheral or periorbital edema, ascites, pleural effusions, and hypertension. Proteinuria is > 3.5 grams per day (on 24-hour urine) and lab tests show hypoalbuminemia and hyperlipidemia

"Compare this to nephritic syndrome which is where there’s peripheral or periorbital edema, hypertension, oliguria, hematuria and proteinuria between 1 and 3 grams per day"
- Primary glomerular diseases mean that the condition occurs on its own, without another known systemic disease such as lupus or diabetes
- Dx is based on the kidney biopsy
- Secondary glomerular diseases are kidney conditions with glomerular pathology in which an underlying cause can be established (DM, HIV, Hep B, Hep C, Lupus, Antiphospholipid syndrome)
Proteinuria occurs because of changes to capillary endothelial cells, the glomerular basement membrane (GBM), or podocytes, which normally filter serum protein selectively by size and charge.
- The disorder results in urinary loss of macromolecular proteins, primarily albumin (protein) but also opsonins, immunoglobulins, erythropoietin, transferrin, hormone-binding proteins, and antithrombin III.
- Massive edema + urine 3.5 grams of protein on 24-hour urine
- Fatty casts with “Maltese cross” sign
- Hypoalbuminemia, hyperlipidemia, and lipiduria
- Oval fat bodies
The most common primary causes are:
Membranous nephropathy: most common in non-diabetic adults associated with malignancies, and infection with hepatitis B virus
- Caused by immune complex formation in the glomerulus - basement membrane becomes damaged
- IgG antibodies target podocyte antigens
Minimal change disease: the most common cause in kids. Assume minimal change disease if a child with idiopathic nephrotic syndrome improves after treatment with corticosteroids
- The cause and pathogenesis of minimal change disease is unclear and it is currently considered idiopathic
Focal segmental glomerulosclerosis (FSGS): obese patients, heroin, and HIV + black males, sickle cell disease
- Podocyte injury or decreased glomerular filtration barrier integrity
- Primary, when no underlying cause is found
- Secondary, when an underlying cause is identified
- Toxins and drugs such as heroin and pamidronate
- Familial forms
- Secondary to nephron loss and hyperfiltration, such as with chronic pyelonephritis and reflux, morbid obesity, diabetes mellitus
The most common secondary causes are:
- Lupus: both nephritic and nephrotic
- Diabetes: a common cause of nephrotic syndrome and subsequent renal failure
- Preeclampsia
Diagnosis is suspected in patients with edema and proteinuria on urinalysis and confirmed by random (spot) urine protein and creatinine levels or 24-h measurement of urinary protein. The cause may be suggested by clinical findings (eg, SLE, preeclampsia, cancer); when the cause is unclear, additional (eg, serologic) testing and renal biopsy are indicated.
- A 24-hour protein collection demonstrating proteinuria above 3.5 grams per day (24-h urine collection) is diagnostic
- Serologic testing and renal biopsy is indicated unless the cause is clinically obvious
- Besides proteinuria, urinalysis may demonstrate casts (hyaline, granular, fatty, waxy, or epithelial cell)
- Lipiduria, the presence of free lipid or lipid within tubular cells (oval fat bodies), within casts (fatty casts), or as free globules, suggests a glomerular disorder causing nephrotic syndrome
- Hypoalbuminemia - serum albumin often is < 3.5 g/dL
- Hyperlipidemia - levels of low-density lipoprotein or LDL above 130 milligrams per deciliter and levels of triglycerides above 150 milligrams per deciliter
Treat the causative disorder and with angiotensin inhibition, Na restriction, and often diuretics and/or statins.
- Minimal change disease: Prednisone 1 mg/kg (up to 80 mg every day) × 4-8 weeks; gradually taper if a response is noted. ACE-Is may also be used as an adjunct to therapy (to reduce proteinuria), or as sole therapy in mild cases.
- Frequent relapsers: Prolonged treatment with immunosuppressants chlorambucil, cyclosporine, or cyclophosphamide should be considered.
- Membranous nephropathy: Depends on the risk of progression to ESRD; judged by the amount of proteinuria and the degree of renal insufficiency. Patients at moderate to high risk should be treated with a combination of glucocorticoids and cytotoxic therapy (cyclophosphamide). Those at low risk can be treated with ACE-Is. Lipid-lowering agents should be used in cases of persistent nephrotic syndrome.
- Focal segmental glomerulosclerosis: ACE-I should be used for the reduction of proteinuria. Immunosuppression with prednisone is the first-line therapy.
- Steroid-resistant cases may benefit from the addition of cyclosporine. Relapses require reinitiation of steroids.
 Osmosis Osmosis |
|
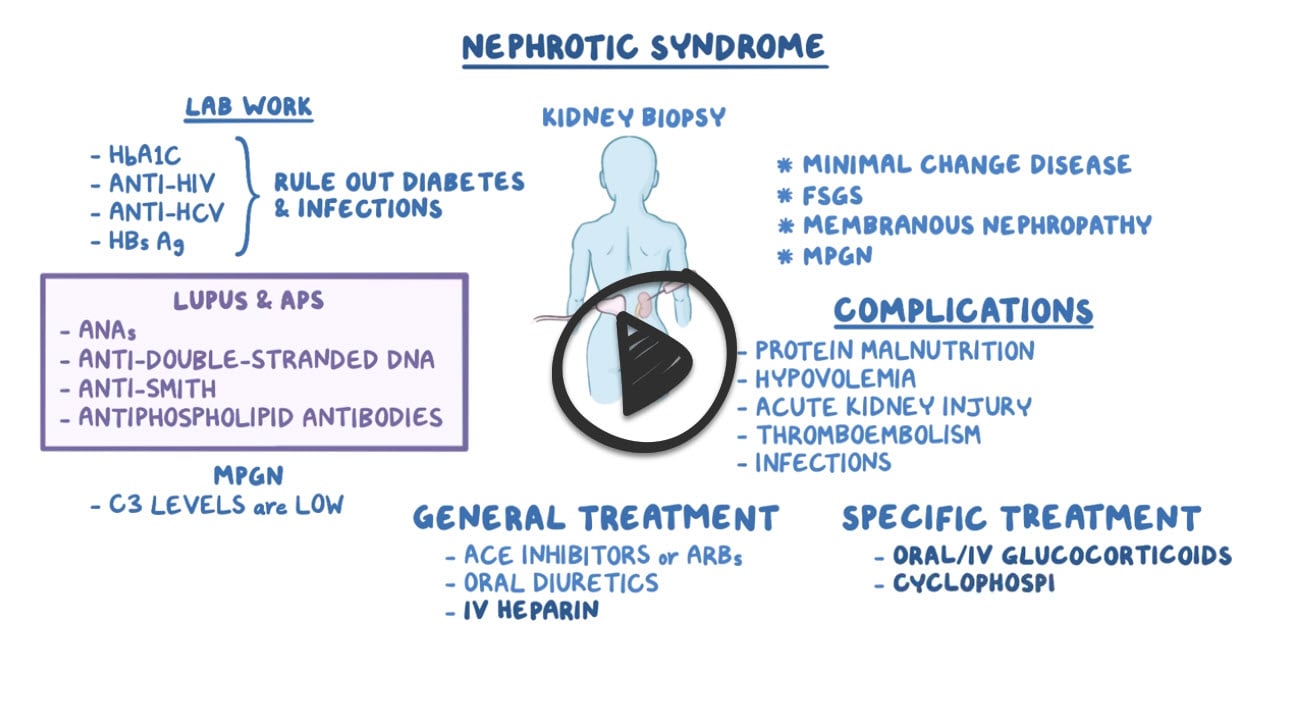 |
 Nephrotic syndrome is a group of symptoms including massive proteinuria defined as a daily loss of 3.5 gm or more of protein, hyperlipidemia, generalized edema, and hypoalbuminemia which results from renal pathology. Nephrotic syndrome is caused by several diseases including membranous glomerulonephritis, minimal change disease, and focal segmental glomerulosclerosis. Nephrotic syndrome is usually initially related to a derangement in the glomerular capillary walls that result in increased permeability to plasma proteins. Loss of protein leads to hypoalbuminemia beyond the compensatory rate of synthesis in the liver, which contributes to generalized edema due to decreased colloid osmotic pressure in the blood. Additionally, nephrotic syndromes are often characterized by immunodeficiency due to loss of immunoglobulins and thrombotic complications due to loss of anticoagulants like antithrombin, protein C and protein S in the urine.
Nephrotic syndrome is a group of symptoms including massive proteinuria defined as a daily loss of 3.5 gm or more of protein, hyperlipidemia, generalized edema, and hypoalbuminemia which results from renal pathology. Nephrotic syndrome is caused by several diseases including membranous glomerulonephritis, minimal change disease, and focal segmental glomerulosclerosis. Nephrotic syndrome is usually initially related to a derangement in the glomerular capillary walls that result in increased permeability to plasma proteins. Loss of protein leads to hypoalbuminemia beyond the compensatory rate of synthesis in the liver, which contributes to generalized edema due to decreased colloid osmotic pressure in the blood. Additionally, nephrotic syndromes are often characterized by immunodeficiency due to loss of immunoglobulins and thrombotic complications due to loss of anticoagulants like antithrombin, protein C and protein S in the urine.
Question 1 |
congestive heart failure Hint: Dependent edema is the most typical finding with CHF. Laboratory findings do not generally include proteinuria or hypoalbuminemia. | |
end-stage liver disease Hint: Symptoms of end-stage liver disease usually include increased abdominal girth indicating ascites. Hypoalbuminemia can occur as a result of malnutrition or concurrently with nephrotic syndrome. | |
nephrotic syndrome | |
malnutrition Hint: Malnutrition is marked by physical wasting, not edema. Hypoalbuminemia may be seen, but hyperlipidemia is not typical. |
Question 2 |
Renal ultrasound Hint: Renal ultrasound may identify hydronephrosis from a stone or other source of obstruction. | |
Renal biopsy | |
Cystoscopy Hint: Cystoscopy can be used in the evaluation of hematuria to assess for bladder or urethral neoplasm, benign prostatic hyperplasia, and radiation or chemical cystitis. | |
Computed tomography scan Hint: CT scanning may identify neoplasms of the kidney or ureter as well as benign conditions such as urolithiasis. |
Question 3 |
Sjögren syndrome
| |
Cushing disease
| |
Hemolytic anemia
| |
Amyloidosis
|
- Minimal-change nephropathy
- Focal glomerulosclerosis
- Membranous nephropathy
- Hereditary nephropathies
- Diabetes mellitus
- Lupus erythematosus
- Viral infections (eg, hepatitis B, hepatitis C, HIV)
- Amyloidosis and paraproteinemias
- Preeclampsia Alloantibodies from enzyme replacement therapy
Question 4 |
Hematuria is among the most common presenting symptoms in adults with nephrotic syndrome
| |
The first sign of nephrotic syndrome in children is usually swelling of the face
| |
The presence of deep venous thrombosis or pulmonary embolism suggests a diagnosis other than nephrotic syndrome
| |
Weight loss and hypotension are frequently present in adults with nephrotic syndrome |
|
List |
References: Merck Manual · UpToDate