
Patient will present as → a 7-year-old boy brought by his parents due to a persistent cough, wheezing, and recurrent lung infections since infancy. The parents also report the child having bulky, foul-smelling stools and difficulty gaining weight despite a good appetite. On examination, he appears underweight, with digital clubbing and crackles heard on lung auscultation. His growth chart shows him below the 3rd percentile for his age. A sweat chloride test shows a chloride level of 88 mmol/L (normal <40 mmol/L). His stool elastase test is low, indicating pancreatic insufficiency. Genetic testing confirms the diagnosis of cystic fibrosis with the identification of mutations in the CFTR gene.
Cystic fibrosis is an autosomal recessive disorder that results in the abnormal production of mucus by almost all exocrine glands, causing obstruction of those glands and ducts.
Although the lungs are generally histologically normal at birth, most patients develop pulmonary disease beginning in infancy or early childhood. Mucus plugging and chronic bacterial infection, accompanied by a pronounced inflammatory response, damage the airways, ultimately leading to bronchiectasis and respiratory insufficiency.
Non-pulmonary - pancreatitis and steatorrhea - patients will need supplementation of fat-soluble vitamins
The course is characterized by episodic exacerbations with infection and progressive decline in pulmonary function.
- Mean survival is about 31 years of age
An elevated quantitative sweat chloride test performed on two different days is diagnostic.
- ↑ NaCL > 60 mEq/L
- However, a normal result does not exclude the diagnosis. If strongly suspected, DNA testing can provide definitive evidence of cystic fibrosis.
- CXR may reveal hyperinflation, mucus plugging, and focal atelectasis
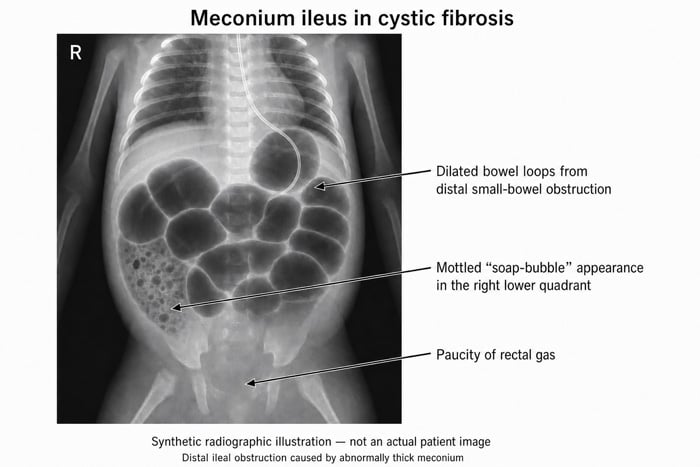
- MECONIUM ILEUS in the newborn — over 90% of infants with meconium ileus have CF. Later: distal intestinal obstruction syndrome (DIOS), rectal prolapse, biliary cirrhosis
***Initially, in the first few months of life, respiratory infection is common with staphylococcus aureus and Haemophilus influenzae
- After that, pseudomonas aeruginosa becomes the major causative organism for infections
All patients with CF should undergo CFTR genotyping to determine if they carry one of the mutations approved for CFTR modulator therapy
- Selection of the CFTR modulator depends upon the CFTR mutation and the child's age
Therapies focus on:
- Clearing the airway of secretions - DNase and/or hypertonic saline and chest physiotherapy
- Reversing bronchoconstriction
- Treating respiratory infections (treat for pseudomonas)
- Replacement of pancreatic enzymes supplement fat-soluble vitamins (A, D, E, K)
- Nutritional and emotional support - high-fat diet
- Routine immunizations: Patients with CF should receive all standard childhood vaccinations
- Seasonal influenza: Annual inactivated flu vaccine for all patients older than 6 months; live attenuated nasal spray not recommended
- Pneumococcal vaccine: Follow CDC guidelines; high-risk children may require additional doses
- COVID-19 vaccine: Strongly recommended for all eligible CF patients
- RSV prophylaxis:
- Nirsevimab: Recommended for infants during the first RSV season; second season for severe lung disease or growth failure
- Palivizumab: Only suggested if nirsevimab is unavailable
 Osmosis Osmosis |
|
 |

Cystic fibrosis is a hereditary disease leading to problems with Cl– channels in the body. It is the most common lethal genetic disease in the Caucasian population. Patients develop recurrent pulmonary infections, bronchitis, infertility, pancreatic insufficiency, steatorrhea, and malabsorption.
Play Video + QuizCystic fibrosis mechanisms

In this Picmonic, Cystic Fibrosis mechanisms are shown as the Sisters with Fibrous-sacks and mechanisms. This disease is inherited in an autosomal recessive manner, the recessive- chocolate, leading to a defect in the CFTR gene on chromosome 7, the CFTR-sifter Chrome 7. This causes a chloride channel defect, the chlorine-dispenser channel broken, which leads to decreased chloride secretion in the gut and lungs, the down-arrow chlorine-dispenser secreting into the Gl and lungs. As a result of decreased chloride secretion, there is increased sodium and water reabsorption intracellularly, shown by the salt-shaker and water-bottle pulled out of the body’s absorbing sponge. Furthermore, as these defective chloride channels cannot reabsorb chloride in sweat glands, patients also have increased sodium and chloride in their sweat, the up-arrow salt-shaker, and chlorine-dispenser at the sweaty-sweat gland. As a result of decreased net water secretion into the Gl and lungs, there is dehydration of mucous layers, the dried-up mucous layers of the body, which is the cause of the specific symptoms in this disease.
Play Video + QuizCystic fibrosis diagnosis and treatment

In this Picmonic, Cystic Fibrosis symptoms and complications are depicted with the sisters with fibrous-sacks with their symptoms and complications. Patients with this disease have recurrent pulmonary infections, the recurrent-clock at the lungs with bacteria, along with chronic bronchitis, the crone broccoli-on-fire. Another association with CF is the development of nasal polyps, the nose polyps. Patients may develop pancreatic insufficiency, the damaged pancreas. Newborns with CF often present with intestinal obstruction, the intestines obstructed by the ship, which is also known as meconium ileus. Malabsorption and diarrhea is not uncommon, the intestinal-mallet and toilet, which can lead to vitamin deficiencies, the Vikings broken. Due to biliary stasis, chronic hepatic disease is often seen, the crone liver. Furthermore, infertility is seen in an overwhelming majority of male patients, the limp male-sign.
Play Video + QuizQuestion 1 |
Positive family history Hint: Cystic fibrosis is a genetic disease, but a positive family history in and of itself is not enough to diagnose the condition. | |
Elevated sweat chloride | |
Recurrent respiratory infections Hint: While recurrent respiratory infections are a classic presentation of cystic fibrosis, the diagnosis relies on confirmation, as noted in explanation B. | |
Elevated trypsinogen levels Hint: Trypsinogen levels are used as a neonatal screening test and if elevated should be followed by more definitive testing to confirm the diagnosis. |
Question 2 |
Diabetic ketoacidosis Hint: While patients with cystic fibrosis most likely will eventually develop insulin-dependent diabetes mellitus, diabetic ketoacidosis is not a common cause of death. | |
Pulmonary infection | |
Intestinal obstruction Hint: While intestinal obstruction may occur in patients with cystic fibrosis, it is not a common cause of death. | |
Acute respiratory failure Hint: See B for explanation. |
Question 3 |
Escherichia coli Hint: See C for explanation.
| |
Staphylococcus epidermidis Hint: See C for explanation.
| |
Pseudomonas aeruginosa | |
Streptococcus pneumoniae Hint: See C for explanation.
|
|
List |
References: Merck Manual · UpToDate